🎨For User
Through this section, you can learn how to upload data and successfully run a workflow. The workflow performs a file format conversion, transforming genomic files from CRAM format to BAM format for downstream analysis.

1. Create Workspace
Name: Only Chinese characters, numbers, letters, “-”, and “” are supported, and the name cannot begin with “-” or “”.
Storage type: The current open-source version only supports NAS storage
Mount directory: Enter the name of the directory to be created in the current NFS. It must begin with “/”.

2. Upload Data
Uploading the data to be analyzed is typically the first step in starting a workflow. In this practice, you need to first upload the required datasets to the NAS directory and manually create a data model table in CSV format. The data files referenced in the data model table must be uploaded to the Bio-OS storage bucket. You can use the links below to download the sample dataset and reference dataset files to your local machine, and then upload them to the NAS storage (using the scp command to copy from local to NAS).
Reference Genome Grch38 dict
Reference Genome Grch38 fasta
Reference Genome Grch38 fai
Sample Data 1(NA12878)
Sample Data 1(Demo)
3. Create Data Model
Click the “+” next to the entity data model to download the CSV template.

In this example, fill in the input or sample file corresponding to each sample in the sample parameter column. Note that the first column field "sample_id" must not be modified.

Once the upload is complete, it should appear as shown in the figure below:

4. Import workflow
Bio-OS uses workflows defined in the Workflow Description Language (WDL) to batch process genomic data. Tools such as GATK, commonly used for processing sample data, typically rely on BAM files as the primary format. While some tools support CRAM, we have observed performance issues when working directly with CRAM files. Therefore, the workflow begins by converting CRAM to BAM.
SAM, BAM, and CRAM are different variants derived from the original SAM format, which was designed to store aligned (or more precisely, mapped) high-throughput sequencing data.
SAM (Sequence Alignment/Map) is a text-based format, formally described in the standard specification.
BAM and CRAM are compressed forms of SAM:
BAM (Binary Alignment/Map) is a lossless compression.
CRAM can range from lossless to lossy compression, depending on the desired level of size reduction (often maximized in practice). Both BAM and CRAM contain the same information and structure as their SAM equivalents; the difference lies in how the files themselves are encoded.
Next, we need to import the Cram-to-Bam workflow into the Workspace:
Click Import Workflow
Enter the corresponding config paramaters
Workflow name: Enter a custom workflow name
Branch/Tag: v0.47
Main path:CramToBam.wdl
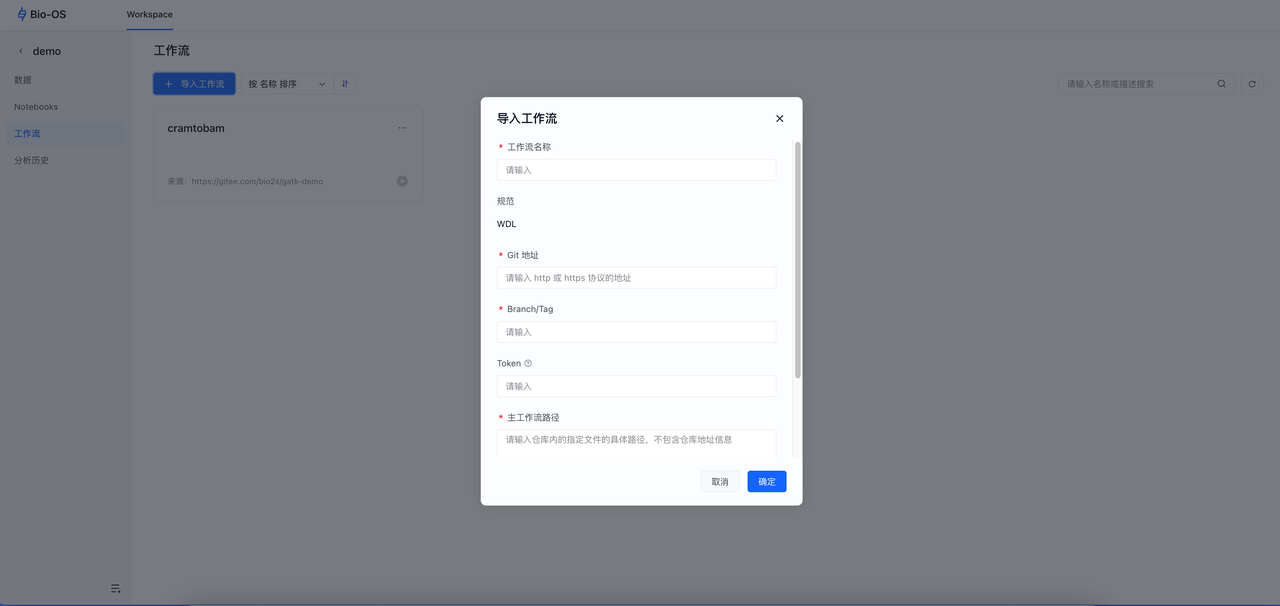
At this point, the Cram-to-Bam conversion workflow has been successfully imported. The WDL source code is provided below: Gitee
5. Submit Workflow
We have just completed the workflow import, and the next step is to run the imported workflow.
Select the Cram-to-Bam workflow, then configure the run options and run parameters.
Choose the data entity uploaded in the first step and specify the corresponding entity data.

Configure input parameters: Select the Input Parameters tab and enter the values as shown below. When there are many input parameters, you can also quickly import them by uploading a JSON file.
Note:The images used in the workflow must support the current architecture. For an arm64 architecture, a compatible image must be used.

Configure output parameters: Use this.bai as the output attribute for the BAI file, and this.bam as the attribute column for the BAM file.
By using this.columnName, the output results are written into the specified column of the selected data table. If the column does not exist in the original table, a new column will be created with that name; if the column already exists, the new results will be filled in or overwrite the existing values.

Click Start Analysis in the upper right corner. The workflow will then be executed by the Cromwell workflow engine, and the results can be viewed in Analysis History.
6.View analysis results
Click Analysis History to view past submissions. In Bio-OS, a submission refers to running batch analyses across multiple samples simultaneously. By clicking Workflow Configuration in the bottom right corner, you can review the workflow configuration of that submission and initiate a new submission from it.

Click the run details of a sample to view the directory where the output files are stored. At this point, you can navigate to the corresponding NAS storage to locate the files.

Last updated